 Chunmei Hu1†
Chunmei Hu1† Yutao Li
Yutao Li Amit Sharma
Amit Sharma Ingo G. H. Schmidt-Wolf
Ingo G. H. Schmidt-Wolf- 1Department of Otolaryngology-Head and Neck Surgery, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
- 2Department of Otolaryngology-Head and Neck Surgery, West China Hospital of Sichuan University, Chengdu, China
- 3Department of Integrated Oncology, Center for Integrated Oncology (CIO), University Hospital Bonn, Bonn, Germany
- 4Department of Neurosurgery, University Hospital Bonn, Bonn, Germany
Head and neck cancer (HNC) ranks as the sixth most prevalent type of cancer globally and accounts for about 4% of all types of cancer. Among all HNC, most are head and neck squamous cell carcinoma (HNSCC) with clinical therapies that include surgery, radiation therapy, chemotherapy, immunotherapy, targeted therapy, and multimodal treatments. In recent years, chimeric antigen receptor (CAR)-T cell immunotherapy has significantly transformed the therapeutic approaches for leukemia and lymphoma and has garnered increased attention as a potential treatment for a wide range of cancers. However, CAR-T immunotherapy in solid tumors, especially HNSCCs, lags significantly behind due to the paucity of tumor-specific antigens, high levels of tumor heterogeneity, immunosuppressive tumor microenvironment, the risk of treatment-related toxicities and off-target adverse events in HNSCCs. The objective of this review is to explore the advancement of CAR-T cell therapy in the treatment of HNSCCs. We aim to outline the targeted antigens in HNSCCs, highlight the challenges and potential solutions, and discuss the relevant combination therapies. Our review presents a comprehensive overview of the recent developments in CAR-T cell therapy for HNSCCs, and provides valuable insights into future research avenues.
1 Introduction
Head and neck cancer (HNC) is a cancer that develops in the facial, oral and neck regions, affecting aerodigestive tract, salivary glands and thyroid. HNC is the seventh most prevalent type of cancer globally (1) and accounts for about 4% of all types of cancer in the United States according to the cancer statistics published in 2021 (2). Tobacco and alcohol consumption are two of the most important risk factors that cause HNC. Human papillomavirus and Epstein-Barr virus infection are also risk factors linked to oropharyngeal cancer and nasopharyngeal cancer, respectively. Among all HNC, most are squamous cell carcinoma that derives from squamous cells of the mucosal epithelium in the facial, oral and neck regions, which are collectively known as HNSCCs. They exhibit immunosuppressive properties and are characterized by the presence of an inflammatory tumor microenvironment (3). Although clinical treatment for HNSCCs includes surgery, radiation therapy, chemotherapy, immunotherapy, targeted therapy, or multimodal therapy, the 5-year survival rate of patients with HNSCC has remained around 50% to 60% over the past few decades (4, 5).
Over the past few years, immunotherapy in cancer treatment has been steadily increasing, with numerous evidence supporting its valuable efficacy. For instance, Food and Drug Administration (FDA) granted approval for the use of Pembrolizumab, a humanized anti-programmed death receptor 1 (PD-1) antibody for the treatment of some patients with an advanced form of head and neck cancer in 2016 (6) whereas cetuximab, a monoclonal antibody that binds competitively to the EGFR, was approved by the FDA for late-stage (metastatic) and resistant cases of squamous cell carcinoma (SCC) of the head and neck in 2011 (7). Apart from PD-1, other immune checkpoints such as cytotoxic T-lymphocyte protein 4 (CTLA4), T-cell immunoreceptor with Ig and ITIM domains (TIGIT), and lymphocyte activation gene 3 protein (LAG3) are considered new rational therapeutic targets (8, 9). In addition, oncolytic viruses (OVs), antigenic vaccines, adoptive T cell transfer (ACT), costimulatory agonists therapy, are also under development (10). Although treatments for HNSCCs have improved consistently over the years, the prognosis of HNSCCs remains unsatisfactory, especially for recurrent and metastatic HNSCCs. Therefore, new treatments that can improve the chance of a complete cure and the prognosis of patients with HNSCCs are required.
Chimeric antigen receptor T (CAR-T) cell therapy is a promising form of immunotherapy which has advanced to commercial approval for application in treating leukemia and lymphoma. To generate CAR-T cells, T cells obtained from either autologous or allogeneic T cells need to be modified to express a CAR (11, 12). With ongoing advancements in CAR-T structural design, CAR-T cells have progressed from their initial first generation to the current fifth generation (13). Typically, a CAR consists of mainly four components: (1) an antigen-binding domain, which is a single-chain variable antibody fragment that is in charge of recognizing tumor antigens (This is not the case for all CARs; for example, the CAR used in T4 immunotherapy does not have this domain); (2) a hinge domain, an extracellular structural segment that allows the extension of the antigen-binding domain from the transmembrane domain. Hinge domains help binding units gain access to target epitopes; (3) a transmembrane domain, which combines extracellular (antigen-binding domain and hinge domain) and intracellular structural domains; (4) an intracellular signaling domain, the part that has changed the most as CAR-T structural designing evolves. This domain encompasses both the signaling and co-stimulatory domain (the presence of the co-stimulatory domain is exclusive to the second and subsequent generations). Intracellular costimulatory molecular are often derived from receptors such as CD28, OX40, and CD137 (14, 15). CAR-T cells behave similarly to T cells, but CAR-T cells are capable of identifying and binding to specific targeted tumor associated antigens (TAAs) or tumor specific antigens (TSAs) on the surface of tumor cells through their specific antigen-binding domains, which initiates anti-tumor immune responses (Figure 1). Apart from this, CAR-T therapy offers a significant advantage over traditional immunotherapy due to its independence from MHC restriction, as immune evasion by cancer cells is mainly caused by a lack of MHC-associated antigen presentation (16). With the encouraging results of CAR-T therapy in treating hematological malignancies, more and more researchers are beginning to explore potential effects of this novel immunotherapy beyond solid tumors, including HNSCCs. Our work will review recent advancements of CAR-T therapy applied in HNSCCs, focusing on the targets of CAR-T cells, the challenges faced, and the corresponding engineering of CARs. Additionally, we will discuss the prospects for combination therapies to further enhance the efficacy of CAR-T therapy in the treatment of HNSCCs.
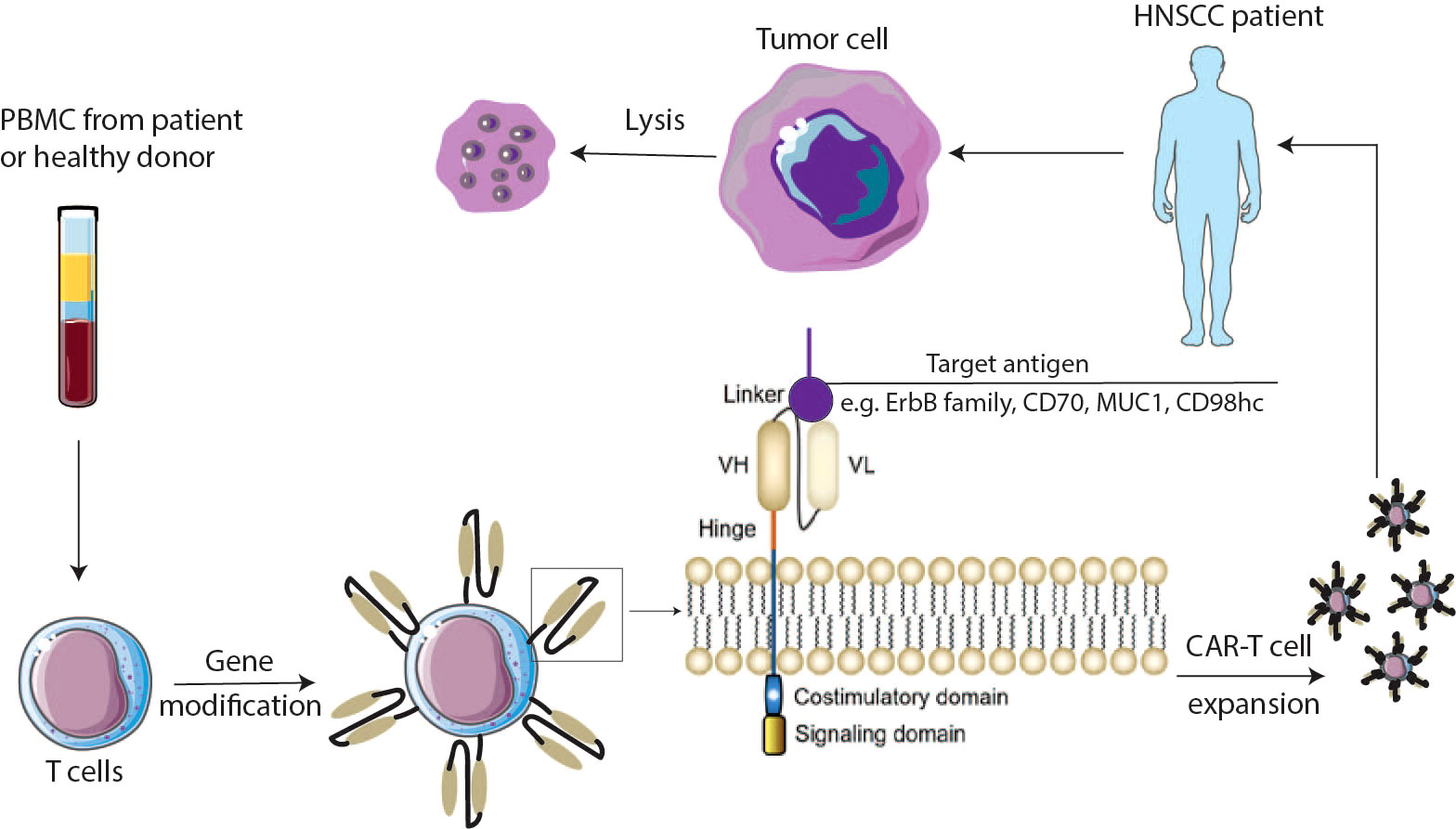
Figure 1 Schematic overview of CAR-T cells. PBMC cells are isolated from the patient’s or donor’s blood and expanded to T cells. Cells are genetically modified to express chimeric antigen receptors (CARs). CAR-T cells are expanded and injected into the HNSCCs patient’s body. CARs are composed of an antigen binding domain which is mainly derived from antibody, a hinge domain bridging between the antigen binding domain and the transmembrane domain, and the intracellular domains including costimulatory and signaling domains. VH—variable heavy chain; VL—variable light chain. CAR chimeric antigen receptor.
2 Target antigens of CAR-T cell therapy in HNSCCs
2.1 ErbB family
Among the potential targets for CAR-T therapy in HNSCCs, the ErbB family (also known as the epidermal growth factor receptor (EGFR) family), holds significant promise. This family is composed of EGFR, Human Epidermal Growth Factor Receptor 2 (HER2 or ErbB2), ErbB3 (HER3), and ErbB4 (HER4) (17, 18). All these receptors can be activated by binding to specific ligands. Upon this, the receptors form homo- or hetero-dimers, which activates their intrinsic tyrosine kinase activity and lead to multiple downstream signaling pathways that play a critical role in regulating cell growth, differentiation, survival, and migration.
The cytotoxic function of CAR-T cells targeted to EGFR against hypopharyngeal squamous cell carcinoma was verified as well in a preclinical study conducted by a team which had already successfully built and verified EGFR-CAR-T-cells (19). This research indicated a notable increase in cytokine secretion following the incubation of EGFR-CAR-T-cells with target tumor cells. This finding not only demonstrated the functional activity of these engineered T cells but also highlighted the enhanced cytotoxic effect specially against FaDu cells, a hypopharyngeal squamous cell carcinoma cell line. The EGFR-CAR-T cells exhibited a target cell lysis rate of 52.66%, further underscoring their potent therapeutic potential of treating HNSCCs.
In addition, T4 immunotherapy, a gene-modified immune cell treatment that specifically targets the ErbB homo- and heterodimers, has been suggested as a solution to the problems that CAR-T cell therapy encounters when treating solid tumors (20, 21). This immunotherapy is recommended because ErbB family members contributed significantly to the development of various types of cancer, including HNSCCs, and selectively targeting ErbB family members using monoclonal antibody therapy exhibits beneficial curative properties (22). It describes the engineering of patient T-cells by retroviral transduction to co-express a chimeric antigen receptor named T1E28z and a chimeric cytokine receptor named 4αβ (17). The T1E28z CAR has a promiscuous ligand of ErbB termed T1E that is connected to CD3ζ through a CD28 hinge/transmembrane and intracellular domain. Being positioned upstream of the CD28+ CD3ζ domain, T1E28z confers specificity on T cells against all EGFR homo- and heterodimers, the ErbB 2/3 heterodimer and all ErbB4 homo- and heterodimers. 4αβ chimeric cytokine receptor is created by fusing the ectodomain of IL-4 cytokine to the β chain used by IL-2 and IL-15. An ex vivo expansion system of CAR-T cells developed by Wilkie et al. suggested that a potent method for rapid proliferation of CAR-T cells is to cultivate them with IL-4 over a period of 7–10 days (23). One of the most challenging barriers of applying T4 immunotherapy is on-target off-tumor toxicity as ErbB family members are expressed in many healthy tissues. Pre-clinical evidence showed a possibility of reducing this toxicity by using the intra-tumoral route and a phase-I clinical study that recruited 13 patients who had been diagnosed with advanced HNSCCs has been developed and demonstrated the safety of intra-tumoral T4 (24). The whole trial is still under recruitment and expected to be completed by 2023 (NCT01818323) (25).
2.2 CD70
Although CD70 expression was high in 19% of HNSCC tumor biopsies, Park et al. utilized a retroviral human CD70 CAR construct to generate CD70 CAR-T and found that it presented the capacity of efficiently eliminating CD70-positive HNSCC cells, which might be related to the transduction efficiency, the CD4+/CD8+ T cell ratio, and a different response to IL-2 stimulation. Consistently, the infusion of CD70-targeted CAR-T cells into patients with clear-cell renal carcinoma showed an outcome of a disease control rate of 76.9%. Among the 14 patients enrolled, one patient had a long-lasting full remission that is still going on as of their 2-year follow-up (26). This outcome further encourages the feasibility of CD70-targeted CAR-T therapy for CD70-positive HNSCC patients.
2.2 MUC1
The MUC1 gene was discovered in human breast cancer with high expression. It is expressed in barrier epithelia lining (27) and skin, immune cells and reproductive organs (28). Therefore, it plays multiple roles in protecting barrier epithelia as well as species propagation in mammals (27). It has been reported that CAR-T cells targeting the tumor MUC1 inhibit triple-negative breast cancer growth in murine models whereas there is minimal damage to normal breast epithelial cells (29). Furthermore, it has been demonstrated that MUC1 has a higher expression in HNSCCs than in adjacent normal tissues in a multivariate analysis (30). Since there was evidence that IL-22 contributed to inducing an elevation of MUC1 expression in colon cancer patients (31), Mei et al. constructed MUC1‐IL22 CAR-T cells and found they exerted more effective cytotoxic function than MUC1 CAR-T cells in vitro and in vivo (3).
2.3 CD98hc
CD98hc, which is a strong related to T cell activation, has been proved to be highly expressed on the surface of radioresistant HNSCC cells (32). CD98hc-targeted UniCAR-T cell was developed and showed ideal ability of tumor cell elimination in a 3D setting of HNSCCs tumor spheroid (33). In addition, the infiltration capability of the UniCAR T cells was improved in the presence of the CD98hc targeted system and a synergistic effect was observed after treatment with fractionated irradiation followed by CD98hc-redirected UniCAR-T delivery.
Apart from these published results, several clinical trials of CAR-T therapy for the treatment of patients with head and neck cancer are underway, targeting novel antigens such as EpCAM, LAMP1, PSMA. We summarized in Table 1.
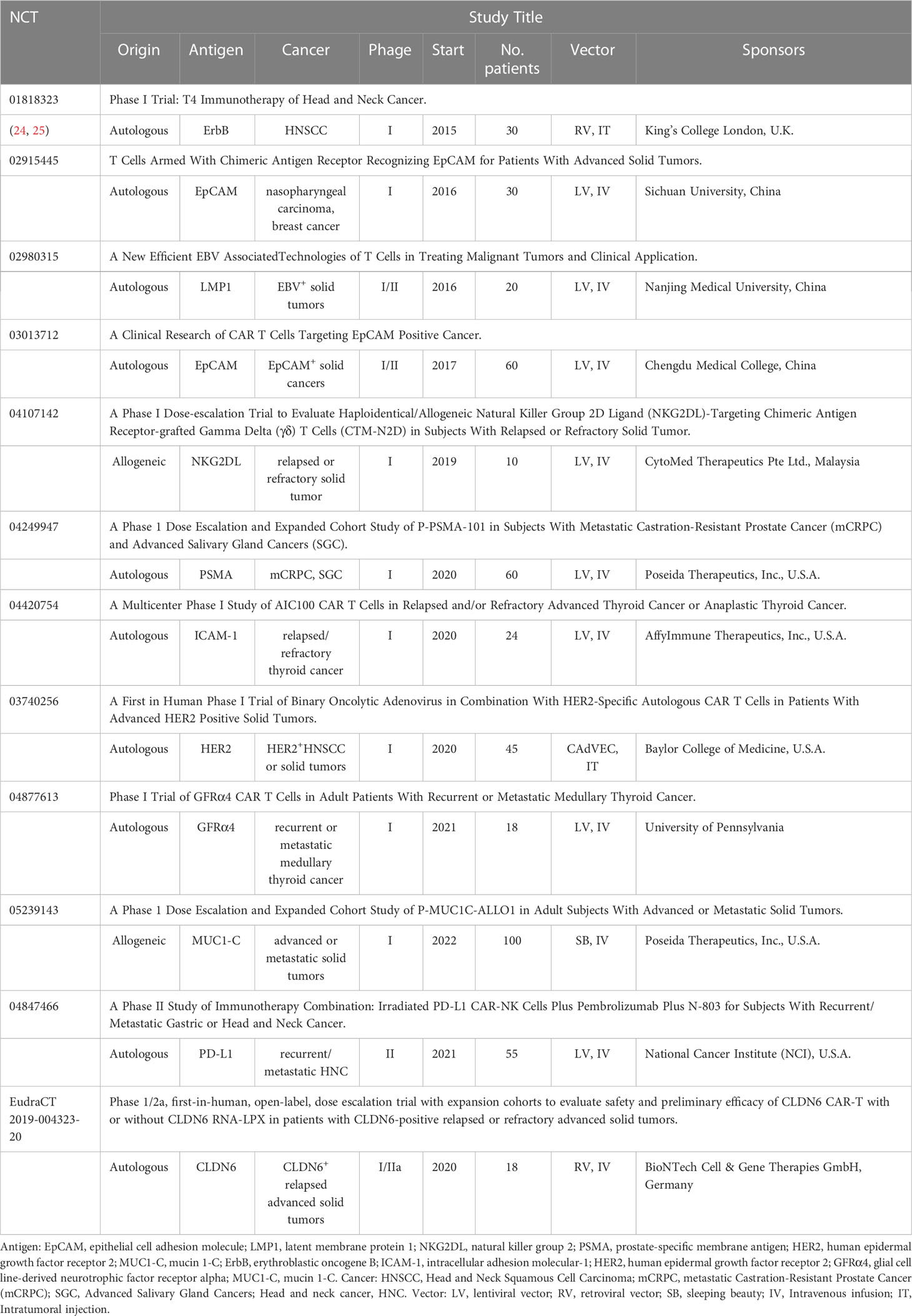
Table 1 Summary of ongoing CAR-T therapy with different target antigens for head and neck cancer on US Clinical Trials Register (clinicaltrials.gov) or EU (clinicaltrialsregister.eu).
3 Overview of challenges of CAR-T cell immunotherapy in the treatment of HNSCCs
3.1 Paucity of tumor specific antigens and high levels of tumour heterogeneity in HNSCCs
Human tumor antigens can be classified into viral antigens, neoantigens that are derived from new, mutated genomic sequences, tumor-specific antigens (TSAs) that derive from unmutated proteins and that are either unique or specific for tumors, and tumor-associated antigens (TAAs) that are differentially expressed in tumor cells and less so in normal cells (34). The optimal target antigens for CAR-T therapy are those tumor-specific antigens (TSAs) exclusively expressed on malignant cells. Specific tumor-specific antigens (TSAs) are neoantigens presented on major histocompatibility complex (MHC) molecules that are suitable for personalized cancer vaccines instead of general CAR-T cell therapy (35). However, these antigens are highly heterogeneous among patients suffering from the same type of tumors, which causes challenges for CAR-T cell therapy. Schumacher, et al. suggest cancer exome-based identification of neoantigens for each patient and assess T cell recognition before generating CAR-T cells (36). Compared to hematological malignancies, targeting TAAs in solid tumors elicits stronger on-target off-tumor effects (37, 38).
When these two antigens can`t achieve satisfactory outcomes in vitro, other options for choosing target antigens for CAR-T might be considered. One alternative antigen is fibroblast activation protein (FAP) in HNSCCs. A novel strategy of CD40 agonist targeted to FAPα combined with radiotherapy has been proven to remodel the TME, activate the CD8+ T cells without the systemic toxicity in murine HPV-positive head and neck tumors. This model increased the overall survival and induced long-term antitumor immunity (39). A previous phase I clinical trial of malignant pleural mesothelioma locally treated with autologous anti-FAP-targeted CAR T-cells (NCT01722149) has been reported that no significant toxicity was observed and the activity of the patient’s redirected T cells was confirmed in vitro (40). Therefore, FAP-targeted CAR-T cells combined with other immune therapies could be a potential strategy for HNSCC patients.
3.2 Inefficient CAR-T cell trafficking in the treatment of HNSCCs
The access of CARs to tumors and their infiltration into the tumor microenvironment (TME) are prerequisites for successful therapies. As with other solid tumors, treatment of HNSCCs must overcome two main barriers: the vascular and stromal barriers. Remodeling of the vascular system hinders immune cells’ ability to penetrate blood vessels to reach the tumor site (41). In the meantime, the stromal barrier of solid tumors consisting of abnormal extracellular matrix (ECM) or mismatched chemokine receptors of immune cells and tumor-derived chemokines also blocks T cell migration (35). Cancer-associated fibroblasts (CAF) within the TME facilitate the accumulation of abnormal ECM around the tumor, eventually creating a compact fibrotic environment. Activating TGF-β can trigger CAF to release extra ECM proteins, limiting T cell’s mobility. Therefore, researchers suppress TGF-β signaling and CAF differentiation, reducing EMC protein secretion and eventually encouraging immune cell infiltration into tumor tissues by inhibiting NOX4, a downstream molecule of TGF-β signaling (42). In the study of Trastuzumab-derived HER2-specific CARs for treating trastuzumab-resistant breast cancer, CAR-T cells can infiltrate the tumor matrix and eliminate tumors that are not accessible to antibodies (43).
In addition, Heparanase (HPSE) can degrade heparin sulfate proteoglycan in the ECM. CAR-T cells expressing HPSE were therefore designed to decompose the ECM and improve T cell infiltration and antitumor efficacy (44). In order to enhance CAR-T cell trafficking, direct injection of CAR-T cells into the tumor site could prevent cells’ circulation through the vascular transport system. For instance, in a study of IL13Rα2-targeted CAR-T cells against glioblastoma, local intracranial delivery of CAR T cells displayed superior anti-tumor efficacy as compared to intravenous administration (45). A similar scenario was reported in murine models with atypical teratoid/rhabdoid tumors (ATRTs) in the treatment with B7-H3-targeted CAR-T cells either intracerebroventricularly (ICV) or intratumorally (IT) compared to intravenously (IV) (46). Even though the lack of clinical lymphodepletion in murine models and the functional distinction between murine and human B7-H3, evidence of intratumaral delivery of CAR-T from animal experiments might provide fundamental data to support clinical trials.
A novel approach for regional delivery of CAR-T cells to solid tumors has also been reported when using implantable biopolymer scaffolds to eliminate heterogeneous tumors. The concurrent transfer of CAR-T cells and the stimulator of INF genes (STING) agonist has the potential to enhance the immune response to tumor cells that do not express the CAR-specific target molecule while locally providing growth factors to CAR-T cells can protect them from the hostile TME during the initial phase of tumor priming (47).
3.3 Immunosuppressive tumor microenvironment in the treatment of HNSCCs
Various immunosuppressive substances and cells that decrease the elicited antitumor immunity are produced and recruited in TME by cancer cells, cancer-associated fibroblasts, and stromal cells (48). In a previous study, the locomotion of CAR-T cells in the solid tumor microenvironment was carefully addressed (49).
Local delivery of cytokines and chemokines by CAR-T cells within the TME, such as IL-22, IL-18, IL-12, and the combination of CCL19 and IL-7, or checkpoint blocking agents, can help in overcoming barriers to T cell infiltration and functioning (50). Besides the exogenous addition of IL-22, CAR-MUC1-IL22 T cells, the fourth-generation CAR-T cells that could release the IL-22 cytokine, may also boost recognition and cytotoxic potential against HNSCC (3).
T-cell exhaustion is significantly influenced by the activation of the transcription factor family NR4A, which is connected to the production of immunosuppressive molecules in solid tumors such as PD-1 and TIM3. CAR-T cells can exhibit better tumor-killing activity and enhanced persistence with the knockout of NR4A (51).
3.4 Treatment-related toxicities in the treatment of HNSCCs
Toxicities associated with CAR-T cell therapy can be classified into two major categories. (1) “On-target off-tumor” toxicity results from specific interactions between the CAR and its target antigen expressed by non-malignant cells. (2) general toxicities, which are related to T cell activation and subsequent systemic release of high levels of cytokines, including cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), hemophagocytic lymph histiocytosis (HLH), and/or macrophage activation syndrome (MAS). ICANS and CRS are the two most severe and unanticipated reactions to CAR-T cell therapy within the category of general toxicities, as reported in the literature (52). CRS, a systemic inflammatory response, is the most common adverse effect caused by CAR-T cell therapy. The most prevalent symptom of CRS following CAR-T cell infusion is fever, which can be accompanied by nausea, fatigue, hypotension, and cardiac dysfunction (53). ICANS is linked with the disturbance of the blood-brain barrier and the rise of cytokine levels in the cerebrospinal fluid. Usually, ICANS arise alongside or after CRS (54). The clinical signs of ICANS linked to CAR-T cell therapy cover encephalopathy, memory impairment, seizures, speech impairment, tremors, headaches, language disturbance, and motor weakness (55, 56). In contrast to CRS, the exact underlying pathophysiology of ICANS is currently vaguely comprehended (57).
Several strategies have been developed to address the challenge of toxicity, such as multitarget CARs, affinity-optimized CARs, inhibitory CARs (iCARs), incorporation of suicide genes into CAR-T cells, and utilization of transient RNA-expressing CARs (58). Kosti et al. developed a CAR-T cell capable of detecting hypoxia. This approach relies on the expression of pan-ErbB-targeted CARs in the hypoxic site. The results demonstrate that CAR molecule expression can only be detected in the hypoxic tumor site of HNSCC, while it is undetectable in normal tissues (59).
4 Therapeutic strategies to enhance CAR-T cells
4.1 Combinatorial antigen targeting
4.1.1 Multiple CARs with “OR” strategy
“OR” strategy refers to immunotherapies with multiple CARs that could be (1) Combined CAR-T cell immunotherapy, which refers to the simultaneous or sequential use of two or more CAR-T cells to treat a similar malignant tumor. This pooled mixture of two populations of CAR-T cells is also referred to as CARpool, (2) co-expression of two to three distinct CARs in individual T cells (dual CAR, triple CAR), or (3) tandem CARs (TanCAR) which are formed by two different scFvs linked together. The “OR” strategy boosts the density of the targetable molecules on the tumor surface, thereby augmenting CAR-T cell potency and antitumor efficacy. For instance, the effective combinatorial application of CAR-CD19-CD28-T cells and CAR-CD19-4-1BB-T cells demonstrates how the Trogocytosis of CAR-T cells may be counteracted with the “OR” Strategy through the CARpool method (60). Nanobodies, heavy chain antibodies found naturally in sharks and camelids, have recently been suggested as scFv substitutes and may enable the production of compact CARs that possess dual specificity and predetermined affinity (61).
4.1.2 CAR-T cells secreting bispecific t-cell engagers (BiTEs)
CAR-T cells can be designed by endogenous, non-engineered T cells through the secretion of BiTEs, which consist of two scFvs, one specific to CD3 and the other to a TAA, connected by a flexible linker. Studies suggest that BiTE-secreting CAR T cells can target multiple antigens, possess better antitumor activity than single-target CAR-T cells, and can prevent tumor escape resulting from antigen heterogeneity while activating and recruiting bystander T cells (42, 62–65). Hence, BiTE-secreting CAR T cells could be a promising therapeutic strategy for HNSCCs.
4.1.3 Universal CARs
The fifth-generation CARs also called universal CARs, employ adapter elements as ligands that enable the targeting of multiple antigens with a distinct CAR-T cell population. Besides, the SUPRA (split, universal, and programmable) CAR system, which consists of a leucine zipper CAR (zipCAR) and an scFv binding to the leucine zipper (zipFv), is only active when zipFv are present, enabling targeting of multiple antigens as well as reducing potential immunogenicity (66).
4.1.4 Multiple CARs with “AND” strategy
The “AND” strategy enhances the safety of CAR-T cells by reprogramming them to activate only in response to target cells expressing two antigens concurrently, resulting in an increased specificity.
Wilkie et al. first hypothesized that CAR-mediated CD3ζ signaling 1 is responsible for the cytotoxicity and IFN-γ production, while the additional CD28-mediated signal 2 promotes T-cell proliferation and IL-2 production. To test it, dual T cells were engineered to co-express ErbB2 and MUC1 with the signaling domains of CD3ζ and CD28 respectively. To test it, dual T cells were engineered to co-express ErbB2 and MUC1 with the signaling domains of CD3ζ and CD28 respectively. This dual CAR-T displayed ErbB2-dependent cytotoxicity and MUC1 and ErbB2-codependent proliferation. However, production of IL-2 was modest compared to control CAR-T containing CD3ζ and CD28 fused domains (67). These findings demonstrate that dual-target CAR-T with cytotoxic and proliferative functions is achievable in vitro.
In addition, Wittsten et al. and Choe et al. developed the synthetic notch (synNotch) “AND” Gate T Cells, capable of inducing target-gene expression upon detecting a cell surface-bound ligand. SynNotch can identify a TAA and stimulate the production of a CAR, which can then activate T-cells upon identification of a second TAA (68). According to research, using synNotch-CAR T cells could potentially serve as a viable approach for solid tumors, decreasing systemic toxicity compared to constitutive CAR expression, as long as the off-tumor target is not near the tumor cells (69, 70).
CAR-T cells performing AND-NOT logic can mitigate adverse effects on benign cells. The approach employs an inhibitory chimeric antigen receptor (iCAR) that selectively recognizes an antigen presented in healthy tissue, combined with an activating CAR that targets a TAA (71). (Figure 2).
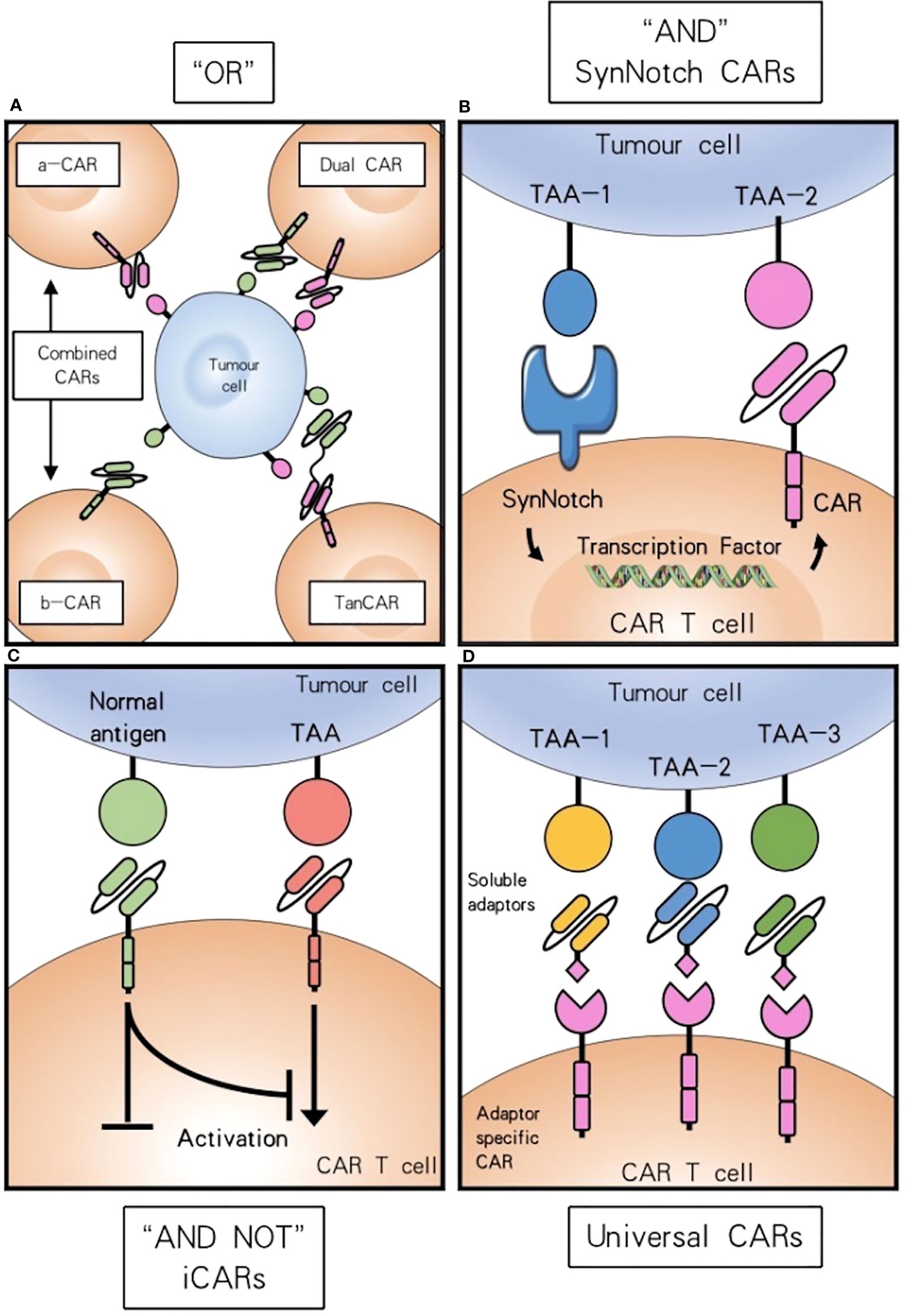
Figure 2 Combinatorial antigen targeting for HNSCCs. (A) For “OR” gate the presence of one antigen is sufficient to trigger effector function, while concurrent expression of both antigens leads to synergistical improvement of activation. (B) “AND” gate SynNotch CAR T cells require the presence of either target antigens to efficiently activate. (C) Combinatorial antigen recognition by AND-NOT logic using an iCAR can increase antigen specificity and safety. (D) Universal CAR T cells can target a variety of different antigens since their antigen specificity comes from the administration of soluble adaptors.
4.2 Increase Success of CAR-T by Chemotherapy and Radiotherapy
The manufacturing process for CAR-T takes about three to four weeks, which can be a fatal waiting time for patients whose disease might continue to be aggressive. Adding bridging chemotherapy prior to CAR-T is currently regarded to be an essential to eradicate tumor cells and boost the efficacy of CAR-T cell therapy in hematologic malignancies (35, 72) Furthermore, the combination of CAR-T therapy with chemotherapy reduces immune suppressor cells in the TME and enhances T cell trafficking to tumors with upregulation of chemokines including CCL5, CXCL9 and CXCL10 (73). However, the combination of chemotherapy with CAR-T cell therapy poses challenges. For instance, the susceptibility of naïve T-cells towards chemotherapy may decrease CAR-T cell efficacy. Besides, chemo-agent cisplatin via HSF1-HSP90 axis enhances the expression of functional PD-L1 in oral squamous cell carcinoma (74). Therefore, the combination effects may be dependent on the dose and the treatment schedule (73).
Radiation therapy (RT) also plays a vital role in the management of HNSCCs and is the primary treatment for nasopharyngeal carcinoma. Radiation modulates tumor immune microenvironments via the release of inflammatory mediators to attract and mediate immune cells. On the other hand, TAAs produced by irradiated tumor cells can be captured by antigen-presenting cells (APCs) (75). Furthermore, activated T cells circle and sequentially have an effect on unirradiated metastases, which is called the abscopal effect (76). With the increasing use of CAR-T, synergistic effects highlight a potential strategy. Further preclinical evidence has been described that NKG2D-based CAR-T cells in mouse glioma models demonstrated efficacy, long-term persistence, and synergistic activity in combination with radiotherapy (77).
Although curative radiotherapy is challenging as a subset of patients are radioresistant (78), combination of conventional radiotherapy with CAR-T therapy turns out encouraging outcomes. Mechanically, RT delivered after CAR T cells resulted in expansion of TCR repertoires, which is an abscopal-like response in a case of a patient with multiply relapsed, refractory myeloma (79). Low-dose RT delivered before CAR T cells supports CD19-targeting CAR T cells in a murine leukemia model (80). In particular, Köseer et al. constructed a 3D culture system of HNSCCs tumor spheroid and demonstrated that the antitumor effect of a CD98hc-retargeted UniCAR-T system was not impaired by irradiation (33). From fractionated irradiation followed by UniCAR-based immunotherapy in Cal33 radioresistant spheroids, they observed that increased UniCAR T expansion with increased EGFP signal and enhanced UniCAR T function with increasing granzyme B and IFN-γ after this combination therapy, indicating an intriguing strategy for the therapy of patients with high-risk HNSCCs (32).
4.3 Improving the efficacy of CAR-T by mRNA vaccine
The feasibility of therapeutic mRNA vaccination against cancer was first demonstrated in 1995 (81). Since then, plenty of studies on mRNA-pulsed DC vaccines have emerged. There are two main mRNA-based immunotherapy approaches. The first strategy is to directly deliver mRNA, involving naked mRNA or mRNA complexed with lipid nanoparticle (LNP), by different administration routes (local injection, such as intramuscular, subcutaneous, and intradermal injection, are major routes for mRNA vaccines in clinical studies). The latter strategy is based on the administration of mRNA-modified DCs or chimeric antigen receptor (CAR) T cells. mRNA vaccines have been utilized in the treatment of aggressive and less accessible solid tumors, however, DC mRNA vaccines are determined by multiple factors. mRNA-encoded proteins are processed to antigenic peptides that associate with MHC class I molecules targeting CD8+ T cells only, not CD4+ T helper cells, to boost antitumor responses. Additionally, posttranscriptional modifications of peptide epitopes (82, 83) and the duration of the antigen peptide-loaded DC presented, DC-cell density, injection site might be considered.
A recent study documented that Reinhard et al. introduced the developmentally regulated tight junction protein claudin 6 (CLDN6) as a CAR target in solid tumors and demonstrated that CAR-T cell-amplifying RNA vaccine was a suitable approach in treating with CLDN6+ lung tumors, without cytokine release syndrome (CRS) (84). Furthermore, this RNA vaccine is efficient in in vivo proliferation and in improving CAR-T cell persistence. Based on data from the Cancer Genome Atlas (TCGA), CLDN6 expression was significantly upregulated in head and neck squamous cell carcinoma (HNSC) (85). Recently, the ongoing clinical trial (EudraCT No. 2019-004323-20) holds a promising future in using CAR-T cell immunotherapy to treat HNSC patients although barriers including high tumor heterogeneity and the maintenance of T cell immune responses in the TME have yet to be addressed.
4.4 Boosting CAR-T by immune checkpoint inhibitors or co-stimulatory molecules
It is well known that immune checkpoint molecules interactions with their ligands prevent recognition between T cells and antigen-presenting cells (86–88). PD-1 and CTLA-4 display distinct functions since they utilize different structural motifs to bind and recruit phosphatases for signal blockade. Blocking of PD-1-PD-L1 but not CTLA-4-B7 interactions enhances tolerized T cell interactions with antigen-bearing DCs and facilitates the phosphorylation of TCR signaling molecules (89). In an orthotopic mouse model of pleural mesothelioma, high doses of both CD28- and 4-1BB-based second-generation CAR T cells eradicated tumors effectively, whereas PD-1 upregulation in the tumor microenvironment inhibited T cell function. Using PD-1 antibody, PD-1 shRNA blockade, or a PD-1 dominant negative receptor restored the function of CD28 CAR-T cells (90). Clinically, a recent phase I clinical study conducted by Adusumilli et al. provided satisfactory median overall survival results for using pembrolizumab and CAR-T together when treating patients with malignant pleural disease (91). An alternative method of targeted delivery of a PD-1-blocking scFv by CAR-T cells not only enhanced the anti-tumor efficacy of CAR-T but also improved bystander tumor-specific T cells in xenogeneic mouse models of PD-L1+ hematologic and solid tumors (92). Clinically, a recruiting phase I trial of T4 immunotherapy of head and neck cancer (NCT01818323) is designed to use panErbB-specific CAR T cells intratumorally and involves the administration of nivolumab. This clinical trial might provide fundamental evidence for the toxicity and persistence of CAR-T combined PD-1 inhibitor. Interestingly, Lee et al. evaluated CD19-targeting CAR-T cells in the context of four different checkpoint combinations-PD-1/TIM-3, PD-1/LAG-3, PD-1/CTLA-4, and PD-1/TIGIT-and found that CAR T cells with PD-1/TIGIT downregulation exerted synergistic antitumor effects while dual downregulation of PD-1/LAG-3 severely damaged antitumor activity. Mechanically, downregulation of PD-1 enhances short-term effector function, whereas downregulation of TIGIT is primarily responsible for remaining a less exhausted state. The efficacy and safety of PD-1/TIGIT-downregulated CD19-targeting CAR-T cells are being assessed in patients with large B cell lymphoma (NCT04836507) (93). The utilization of dual immune checkpoint blockades might provide novel perspectives for the treatment of patients with solid tumors in CAR-T therapy. However, it is vital to carefully monitor the potential risk such as CRS and ICANS.
Conversely, CD80/CD86-targeted CAR-T (CTLA4-CAR), which comprises the extracellular and transmembrane portions of human CTLA4, the cytoplasmic region of human CD28, and the intracellular domains of human CD3z, was designed to destroy CD80/CD86-associated tumor cells in vitro and vivo. CTLA4-CAR T cells promoted IL-2 and IFN-γ secretion and suppressed tumor growth in xenograft models of malignant B cells. Furthermore, CTLA4-CAR T cells were found to infiltrate in residual tumors and are cytotoxic to myeloid-derived suppressor cells (MDSCs) without signs of severe CRS in murine models (94). Similarly, CTLA-4 tail fusion has superior anti-tumor efficacy in a relapsed leukemia model (95).
Apart from immune checkpoints, co-stimulatory members of the TNFRSF molecule (4-1BB, OX40, CD27, CD40, HVEM, and GITR) display unique functional roles in CAR-T therapy for solid tumors. CD40 ligand-modified CAR T cells displayed superior antitumor efficacy, promoted upregulation of co-stimulatory markers CD80 and CD86 on CD40+ lymphoma cells, educated antigen-presenting cells, enhanced recruitment of immune effectors, and mobilized specific tumor-recognizing T cells without outward signs of toxicity (96). Mata et al. introduced an inducible costimulatory (iCO) molecule consisting of a chemical inducer of dimerization (CID)-binding domain and the MyD88 and CD40 signaling domains into CAR-T and found that the CAR-T cells improve the efficacy of CAR T-cell therapy for solid tumors (97). Golubovskaya et al. designed CAR with a glucocorticoid-induced TNFR-related protein (GITR) co-stimulatory domain and recorded that EGFR-GITR-CD3 CAR-T cells were cytotoxic against EGFR+ pancreatic and ovarian cancer cells with higher levels of IFN-γ (98). These results suggest co-stimulatory molecules could be a potential option to enhance the activity of CAR-T in HNSCCs.
4.5 Oncolytic adenoviruses
Oncolytic adenoviruses are one of the most employed oncolytic viruses as they can elicit immune-stimulating signals that can substantially eliminate local immunosuppression, cause tumor cell lysis, and improve antitumor immune responses (99). However, limitations such as short persistence, host antiviral immune response, are impeding the utilization of the clinic (100). To overcome them, helper-dependent adenoviral vectors (HDAds) are deleted of all viral genes and can be armed with approximately 34 kb of therapeutic transgenes, which have demonstrated better efficacy for CAR-T immune therapy (101). Further evidence suggests that the feasibility of encoding the PD-L1 blocking antibody and IL-12p70 (CAd12_PDL1) combined with HER2-CAR-T cells infusion in HNSCC xenograft and orthotopic models. Results showed controlled primary and metastasized tumor growth and prolonged survival (102). In addition to this, a phase I trial involving patients with HER2-positive cancer including HNSCCs, utilizing CAdVEC (a genetically modified oncolytic viral strain of human adenovirus (Ad) with potential immunostimulating and antineoplastic activities) coupled with HER2-specific CAR-T cells, is in recruitment (NCT03740256). We have summarized different strategies to enhance the efficacy of CAR-T cell therapy (Figure 3).
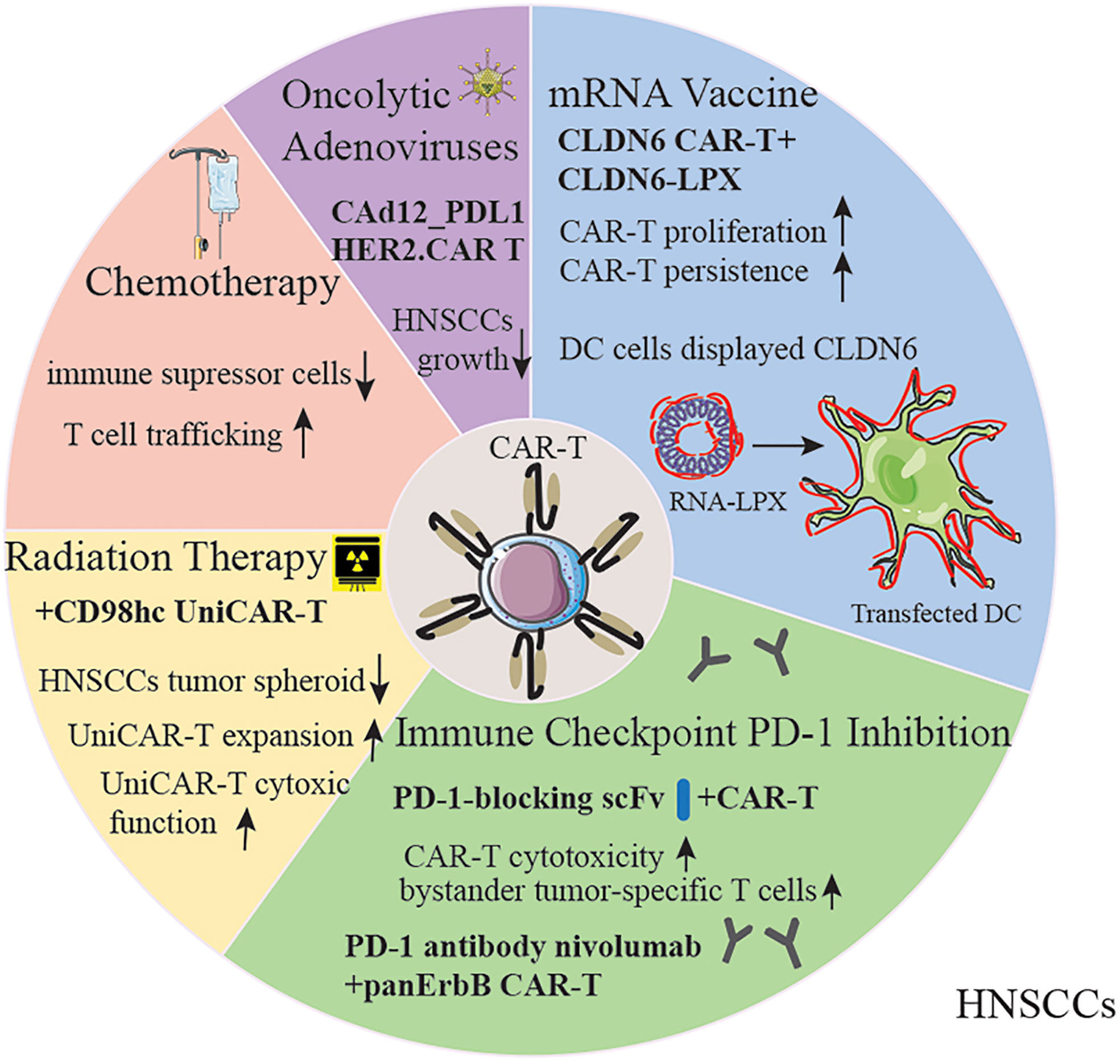
Figure 3 Strategies for improving CAR-T therapy for HNSCC patients. The capability of CAR-T cells could be amplified by chemotherapy, radiation therapy, oncolytic adenoviruses, mRNA vaccine and immune checkpoint inhibitors. DC, Dendritic Cells; RNA-LPX, RNA-lipoplex.
5 Overview of current status of CAR-T clinical trials in HNSCCs
NCT01818323 is the first ongoing clinical trial for patients with locally advanced/recurrent HNSCC. T4 immunotherapy is an autologous T cell therapy. A retroviral vector has been used to modify T cells for gene therapy. T cells coexpress two chimeric receptors: T1E28z and 4αβ. T1E28z is a chimeric antigen receptor that engages multiple ErbB dimers commonly overexpressed in HNSCC. On the other hand, previous evidence demonstrated that the addition of IL-4 to T-cells that express 4αβ induced STAT3/STAT5/ERK phosphorylation and exponential proliferation (23). 4αβ, a chimeric cytokine receptor, is therefore designed to insert into T4+ T-cells (24). T4 immunotherapy displays antitumor activity against HNSCC cell lines and tumors in vivo, without significant toxicity to mice. In this clinical trial, a traditional 3 + 3 dose escalation design is used. If maximum tolerated dose remains undefined, cohorts 6/7 will receive low or high-dose cyclophosphamide before 1x109 T4+ T cells infusion. In cohort 7, patients are designed to receive 3 doses of nivolumab before CAR-T treatment (24). However, risk of on-target off-tumor toxicity should be considered due to low-level ErbB expression in normal tissues. To overcome it, a ErbB-targeted CARs capable of detecting the hypoxia of HNSCC might be an option in the future (59). Further investigation on the feasibility of T4 manufacture was evaluated by Papa et al. The results suggested that T4 manufacture was successful, and the overall disease control rate for patients with advanced HNSCC was 69% after T4 immunotherapy without lymphodepletion. In addition, treatment-related adverse effects were ≤ grade 2, without dose-limiting toxicities. One patient achieved a rapid and complete response after PD-1 inhibitor nivolumab treatment (25) (Figure 4). Therefore, intra-tumoral administration of T4 in patients with advanced HNSCC could be safe and effective. When HNSCC patients are treated with CAR-T cells by multifocal intra-tumoral injection, lymphodepletion might not be necessary. Conversely, Hirayama et al. reported that the response to lymphodepletion impacts progression-free survival (PFS) in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR-T cells. Higher intensities of cyclophosphamide and fludarabine lymphodepletion are associated with higher levels of monocyte chemoattractant protein-1 (MCP-1) and IL-7, contributing to better PFS (NCT01865617) (103). However, the evidence of CAR-T for human solid tumors is rare. Suryadevara et al. revealed that CD28-4-1BBz CARs therapy required lymphodepletion to eliminate Tregs cells in murine solid tumor models (104). Inconsistently, Wang et al. generated PD-1 and T cell receptor (TCR) deficient mesothelin-specific CAR-T (MPTK-CAR-T) cells using CRISPR-Cas9 technology and assessed the efficacy of CAR-T in MSLN+ patients with advanced solid tumors who received a second or third infusion of MPTK-CAR-T cells without prior lymphodepletion. There was no dose-limiting toxicity or unexpected adverse events. Nevertheless, the approach of using CRISPR-engineered T cells to delete TCR in the study cannot be compared to T4 immunotherapy due to the differences in the CAR-T construct, the type of solid tumor and the delivery method. Whether lymphodepletion impacts CAR-T cell expansion, functionality, long-term persistence and toxicity in the treatment of solid tumors should be further investigated (105).
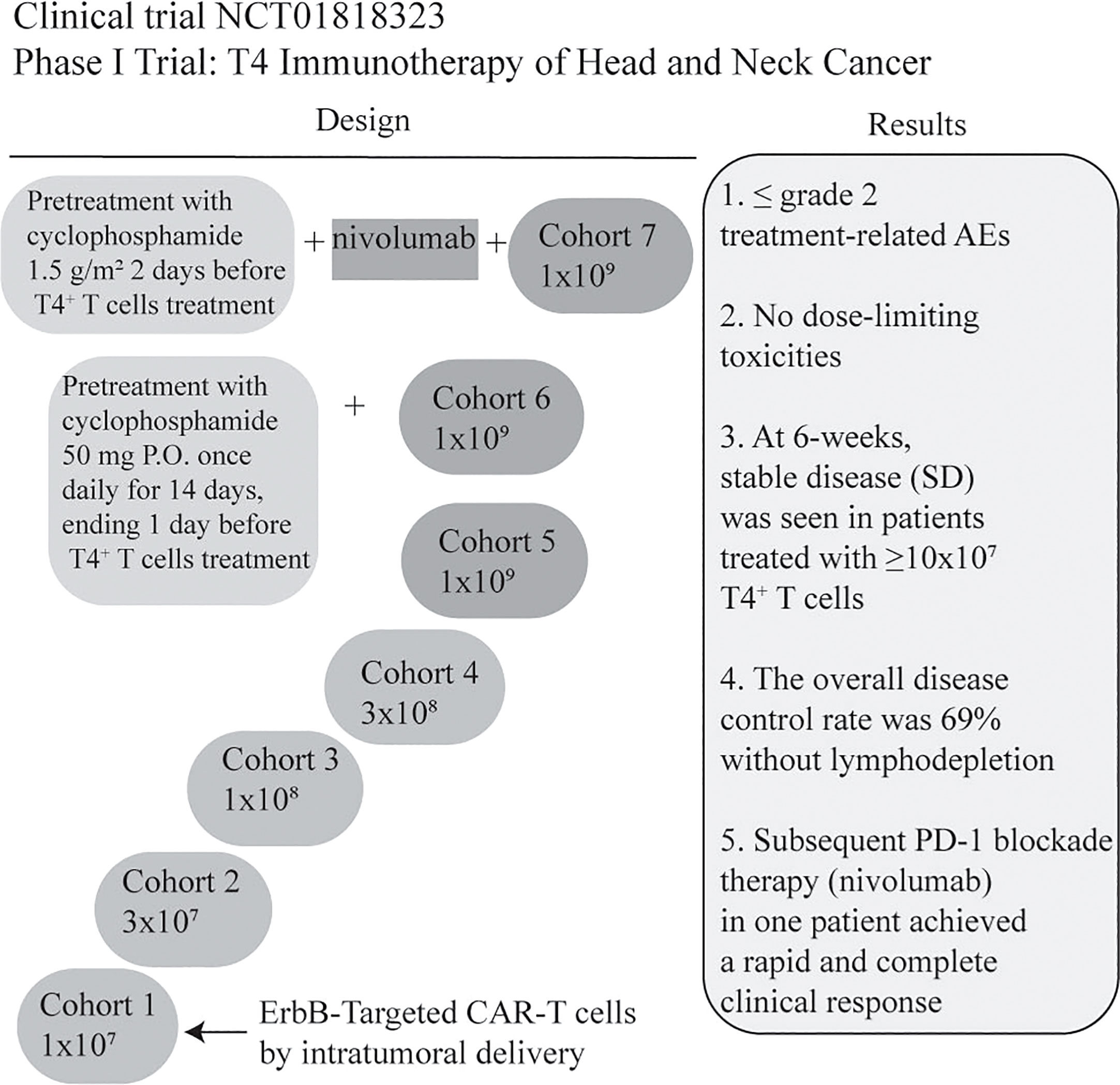
Figure 4 Summary of a Phase I clinical trial of T4 Immunotherapy for head and neck cancer. CAR-T cells co-express T1E28ζ, a promiscuous ErbB ligand coupled to a CD28+CD3ζ endodomain, and 4αβ, an IL-4-responsive chimeric cytokine receptor. CAR-T manufacturing was successful in all cases (13/13) even though the rate of lymphopenia was 62%. The dose of CAR T-cell was escalated through 5 cohorts from 1x107 -1x109 administered as a single treatment, by multifocal intra-tumoral injection without lymphodepletion. All patients will be monitored for dose-limiting toxicities (DLT) for 4 weeks before proceeding to the next dose level (24). In addition, the clinical results are illustrated briefly in the figure (25). Treatment-related AEs: Treatment-related adverse events (AEs); nivolumab: anti-PD-1IgG4 antibody; P.O.: per mouth.
Most current clinical trials targeting different antigens of HNSCC patients are in recruiting status except for NCI 04249947 and EudraCT 2019-004323-20 are ongoing. Among these clinical trials, patients with HNSCC will receive autologous T cell infusion. However, in NCI 01818323 and NCI 03740256, participants will receive intratumoral injection to reduce the toxicity of CAR-T therapy. Additionally, there is a CAR-γδT (NCT04107142) and CAR-NK cell (NCT04847466) trial targeting HNSCC patients that has been in recruiting status.
Notably, in a phase 1/2a clinical trial conducted in Germany, an RNA vaccine was employed by BioNTech Cell & Gene Therapies GmbH in patients with CLDN6+ refractory or relapsed advanced solid tumors (EudraCT No. 2019-004323-20). This study aimed at investigating and assessing the security and initial efficacy of CLDN6 CAR-T cells with or without CLDN6 RNA-lipoplexes (LPX) among the recruited patients. RNA-LPX are composed of liposomes and are widely utilized as in vitro transfection reagents. Briefly, this CAR Vaccine is derived from BioNTech´s RNA-LPX technology modified by delivery nanoparticles to APC or negatively charged mRNA, or by shielding positive surface charges with PEG (106, 107). It is an ongoing, multicenter clinical trial involving in our Integrated Oncology Center (CIO) (Table 1) and might be a promising strategy for HNSCCs immune therapy in the future.
6 Future perspectives
CAR-T cell therapy remains in its infancy although it has been moving beyond its extraordinary success in hematological malignancies. Extensive research in solid tumors revealed that CAR-T cell therapy has tremendous promise in treating refractory and metastatic neoplasms resistant to conventional treatments, including HNSCCs. Recent advances in engineering technologies, novel target antigen spotting, and combination therapies have shown great potential in overcoming hurdles to the development of CAR-T cell therapy in HNSCCs, such as related on-target off-tumor toxicities, the immunosuppressive nature of the tumor microenvironment, and poor T-cell homing to target antigens. Still, further research is necessary before CAR-T cell treatment could be used effectively and safely for people with HNSCCs.
In addition, genetically modifying γδT and NK cells to express CARs has demonstrated encouraging preclinical outcomes and is presently being investigated in a number of clinical studies. More importantly, CIK cells with dual innate and adaptive cytotoxic function have already been modified into CAR-CIK in preclinical solid tumor treatment (108–111) and more effective based on biodistribution and toxicity analyses. Therefore, CAR-CIK might be a new paradigm in solid tumor cellular immunotherapy.
In particular, personalized CAR-T immunotherapy might address the issue of solid tumor heterogeneity. Methodologically, Cafri et al. developed a novel approach using tumor-infiltrating lymphocytes to identify the specific immunogenic mutations expressed in patients’ tumors. They concatenated neoantigens and mutations of driver genes into a single mRNA construct to vaccinate patients with metastatic gastrointestinal cancer. This method provides the possibility of using specific immunogenic mutations in mRNA-LPX or predicted neoepitopes in CAR-T therapy (112). Intriguingly, Miltenyi Biotec B.V. & Co. KG introduced the MICS (MACSima Imaging Cyclic Staining) technology, which identifies hundreds of protein targets derived from a single specimen based on immunofluorescent imaging. Using this technology, we can identify potential target genes derived from individual patients for CAR-T therapy (113). Further advanced methods could be applicable for CAR-T personalized therapy.
Author contributions
CH, ML YL and YZ wrote the manuscript. CH, ML and YL revised the manuscript. CH, HL and YL reviewed the manuscript and AS provided helpful discussions. IS-W revised and supervised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA A Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654
3. Mei Z, Zhang K, Lam AK-Y, Huang J, Qiu F, Qiao B, et al. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med (2020) 9:640–52. doi: 10.1002/cam4.2733
4. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer (2010) 11:9–22. doi: 10.1038/nrc2982
5. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–86. doi: 10.1002/ijc.29210
6. Cohen EEW, Bell RB, Bifulco CB, Burtness B, Gillison ML, Harrington KJ, et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J Immunother Cancer (2019) 7:184. doi: 10.1186/s40425-019-0662-5
7. Stanbouly D, Philipone E, Morlandt AB, Kaleem A, Chuang SK, Patel N. Adverse events secondary to cetuximab therapy in head & neck cancer therapy and risk factors for serious outcomes. Oral Oncol (2022) 131:105952. doi: 10.1016/j.oraloncology.2022.105952
8. Gameiro SF, Ghasemi F, Barrett JW, Koropatnick J, Nichols AC, Mymryk JS, et al. Treatment-naïve HPV+ head and neck cancers display a T-cell-inflamed phenotype distinct from their HPV- counterparts that has implications for immunotherapy. Oncoimmunology (2018) 7:e1498439. doi: 10.1080/2162402X.2018.1498439
9. Eichberger J, Spoerl S, Spanier G, Erber R, Taxis. J, Schuderer J, et al. TIGIT expression on intratumoral lymphocytes correlates with improved prognosis in oral squamous cell carcinoma. Biomedicines (2022) 10:3236. doi: 10.3390/biomedicines10123236
10. Yu C, Li Q, Zhang Y, Wen ZF, Dong H, Mou Y. Current status and perspective of tumor immunotherapy for head and neck squamous cell carcinoma. Front Cell Dev Biol (2022) 10:941750. doi: 10.3389/fcell.2022.941750
11. Vormittag P, Gunn R, Ghorashian S, Veraitch FSA. Guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol (2018) 53:164–81. doi: 10.1016/j.copbio.2018.01.025
12. Chen YJ, Abila B, Kamel YM. CAR-T: what is next? Cancers (Basel) (2023) 15:663. doi: 10.3390/cancers15030663
13. Asmamaw Dejenie T, Tiruneh G, Medhin M, Dessie Terefe G, Tadele Admasu F, Wale Tesega W, et al. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum Vaccines Immunotherapeutics (2022) 18:2114254. doi: 10.1080/21645515.2022.2114254
14. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
15. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
16. Lu J, Jiang G. The journey of CAR-T therapy in hematological malignancies. Mol Cancer (2022) 21:194. doi: 10.1186/s12943-022-01663-0
17. Larcombe-Young D, Papa S, Maher J. PanErbB-targeted CAR T-cell immunotherapy of head and neck cancer. Expert Opin Biol Ther (2020) 20:965–70. doi: 10.1080/14712598.2020.1786531
18. Palumbo C, Benvenuto M, Focaccetti C, Albonici L, Cifaldi L, Rufin A, et al. Recent findings on the impact of ErbB receptors status on prognosis and therapy of head and neck squamous cell carcinoma. Front Med (Lausanne). (2023) 10:1066021. doi: 10.3389/fmed.2023.1066021
19. Dong Y-H, Ding Y-M, Guo W, Huang J-W, Yang Z, Zhang Y, et al. The functional verification of EGFR-CAR T-cells targeted to hypopharyngeal squamous cell carcinoma. ONCOTARGETS AND Ther (2018) 11:7053–9. doi: 10.2147/OTT.S175516
20. Prinzing B, Krenciute G. Hypoxia-inducible CAR expression: an answer to the on-target/off-tumor dilemma? Cell Rep Med (2021) 2:100244. doi: 10.1016/j.xcrm.2021.100244
21. Klampatsa A, Achkova DY, Davies DM, Parente-Pereira AC, Woodman N, Rosekilly J, et al. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett (2017) 393:52–9. doi: 10.1016/j.canlet.2017.02.015
22. Papa S, van Schalkwyk M, Maher J. Clinical evaluation of ErbB-targeted CAR T-cells, following intracavity delivery in patients with ErbB-expressing solid tumors. Methods Mol Biol (2015) 1317:365–82. doi: 10.1007/978-1-4939-2727-2_21
23. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira ACP, Cleary S, van der Stegen SJC, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem (2010) 285:25538–44. doi: 10.1074/jbc.M110.127951
24. van Schalkwyk MCI, Papa SE, Jeannon J-P, Guerrero Urbano T, Spicer JF, Maher J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum Gene Ther Clin Dev (2013) 24:134–42. doi: 10.1089/humc.2013.144
25. Papa S, Adami A, Metoudi M, Achkova D, van Schalkwyk M, Pereira AP, et al. A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC). J OF Clin Oncol (2018) 36:suppl 3046–3046. doi: 10.1200/JCO.2018.36.15_suppl.3046
26. Hemonc today off-the-Shelf CAR-T produces complete remission in patient with advanced kidney cancer . Available at: https://www.healio.com/news/hematology-oncology/20221111/offtheshelf-cart-produces-complete-remission-in-patient-with-advanced-kidney-cancer.
27. Kufe DW. Emergence of MUC1 in mammals for adaptation of barrier epithelia. Cancers (Basel). (2022) 14:4805. doi: 10.3390/cancers14194805
28. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. (2009) 9:874–85. doi: 10.1038/nrc2761
29. Zhou R, Yazdanifar M, Roy LD, Whilding LM, Gavrill A, Maher J, et al. Corrigendum: CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front Immunol (2020) 11:628776. doi: 10.3389/fimmu.2020.628776
30. Rabassa ME, Croce MV, Pereyra A, Segal-Eiras A. MUC1 expression and anti-MUC1 serum immune response in head and neck squamous cell carcinoma (HNSCC): a multivariate analysis. BMC Cancer (2006) 6:253. doi: 10.1186/1471-2407-6-253
31. Noyama Y, Okano M, Fujiwara T, Kariya S, Higaki T, Haruna T, et al. IL-22/IL-22R1 signaling regulates the pathophysiology of chronic rhinosinusitis with nasal polyps via alteration of MUC1 expression. Allergology Int (2017) 66:42–51. doi: 10.1016/j.alit.2016.04.017
32. Digomann D, Kurth I, Tyutyunnykova A, Chen O, Löck S, Gorodetska I, et al. The CD98 heavy chain is a marker and regulator of head and neck squamous cell carcinoma radiosensitivity. Clin Cancer Res (2019) 25:3152–63. doi: 10.1158/1078-0432.CCR-18-2951
33. Köseer AS, Loureiro LR, Jureczek J, Mitwasi N, González Soto KE, Aepler J, et al. Validation of CD98hc as a therapeutic target for a combination of radiation and immunotherapies in head and neck squamous cell carcinoma. Cancers (Basel) (2022) 14:1677. doi: 10.3390/cancers14071677
34. Vigneron N. Human tumor antigens and cancer immunotherapy. BioMed Res Int (2015) 2015:948501. doi: 10.1155/2015/948501
35. Chung H, Jung H, Noh J-Y. Emerging approaches for solid tumor treatment using CAR-T cell therapy. IJMS (2021) 22:12126. doi: 10.3390/ijms222212126
37. Pan K, Farrukh H, Chittepu VCSR, Xu H, Pan C, Zhu Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res (2022) 41:119. doi: 10.1186/s13046-022-02327-z
38. Shang S, Zhao Y, Qian K, Qin Y, Zhang X, Li T, et al. The role of neoantigens in tumor immunotherapy. BioMed Pharmacother. (2022) 151:113118. doi: 10.1016/j.biopha.2022.113118
39. Labiano S, Roh V, Godfroid C, Hiou-Feige A, Romero J, Sum E, et al. CD40 agonist targeted to fibroblast activation protein α synergizes with radiotherapy in murine HPV-positive head and neck tumors. Clin Cancer Res (2021) 27:4054–65. doi: 10.1158/1078-0432.CCR-20-4717
40. Curioni A, Britschgi S, Hiltbrunner L, Bankel P, Gulati WW, Opitz I, et al. A phase I clinical trial of malignant pleural mesothelioma treated with locally delivered autologous anti-FAP-targeted CAR T-cells. Ann Oncol (2019) SUPPLEMENT 5:V501. doi: 10.1093/annonc/mdz253.052
41. Zhang H, Zhu S, Deng W, Li R, Zhou H, Xiong H. The landscape of chimeric antigen receptor T cell therapy in breast cancer: perspectives and outlook. Front Immunol (2022) 13:887471. doi: 10.3389/fimmu.2022.887471
42. Cao G, Zhang G, Liu M, Liu J, Wang Q, Zhu L, et al. GPC3-targeted CAR-T cells secreting B7H3-targeted BiTE exhibit potent cytotoxicity activity against hepatocellular carcinoma cell in the in vitro assay. Biochem Biophys Rep (2022) 31:101324. doi: 10.1016/j.bbrep.2022.101324
43. Szöőr Á., Tóth G, Zsebik B, Szabó V, Eshhar Z, Abken H, et al. Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett (2020) 484:1–8. doi: 10.1016/j.canlet.2020.04.008
44. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med (2015) 21:524–9. doi: 10.1038/nm.3833
45. Brown CE, Aguilar B, Starr R, Yang X, Chang W-C, Weng L, et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther (2018) 26:31–44. doi: 10.1016/j.ymthe.2017.10.002
46. Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-Targeted CAR T cells for treatment of atypical Teratoid/Rhabdoid tumors. Nat Med (2020) 26:712–9. doi: 10.1038/s41591-020-0821-8
47. Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest (2017) 127:2176–91. doi: 10.1172/JCI87624
48. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol (2020) 17:1–12. doi: 10.1038/s41423-019-0306-1
49. Nguyen A, Johanning G, Shi Y. Emerging novel combined CAR-T cell therapies. Cancers (Basel) (2022) 14:1403. doi: 10.3390/cancers14061403
50. Watanabe K, Kuramitsu S, Posey AD, June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol (2018) 9:2486. doi: 10.3389/fimmu.2018.02486
51. Chen J, López-Moyado IF, Seo H, Lio CWJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature (2019) 567:530–4. doi: 10.1038/s41586-019-0985-x
52. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
53. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
54. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
55. Hu Y, Sun J, Wu Z, Yu J, Cui Q, Pu C, et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J Hematol Oncol (2016) 9:70. doi: 10.1186/s13045-016-0299-5
56. Gust J, Hay KA, Hanafi L-A, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discovery (2017) 7:1404–19. doi: 10.1158/2159-8290.CD-17-0698
57. The Lancet Oncology. CAR T-cell therapy for solid tumours. Lancet Oncol (2021) 22:893. doi: 10.1016/S1470-2045(21)00353-3
58. Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer (2019) 18:125. doi: 10.1186/s12943-019-1057-4
59. Kosti P, Opzoomer JW, Larios-Martinez KI, Henley-Smith R, Scudamore CL, Okesola M, et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep Med (2021) 2:100227. doi: 10.1016/j.xcrm.2021.100227
60. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature (2019) 568:112–6. doi: 10.1038/s41586-019-1054-1
61. De Munter S, Ingels J, Goetgeluk G, Bonte S, Pille M, Weening K, et al. Nanobody based dual specific CARs. Int J Mol Sci (2018) 19:E403. doi: 10.3390/ijms19020403
62. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
63. Tatari N, Zhang X, Chafe SC, McKenna D, Lawson KA, Subapanditha M, et al. Dual antigen T cell engagers targeting CA9 as an effective immunotherapeutic modality for targeting CA9 in solid tumors. Front Immunol (2022) 13:905768. doi: 10.3389/fimmu.2022.905768
64. Yin Y, Rodriguez JL, Li N, Thokala R, Nasrallah MP, Hu L, et al. Locally secreted BiTEs complement CAR T cells by enhancing killing of antigen heterogeneous solid tumors. Mol Ther (2022) 30:2537–53. doi: 10.1016/j.ymthe.2022.05.011
65. Okuma A, Ishida Y, Kawara T, Hisada S, Araki S. Secretory Co-factors in next-generation cellular therapies for cancer. Front Immunol (2022) 13:907022. doi: 10.3389/fimmu.2022.907022
66. Miao L, Zhang Z, Ren Z, Tang F, Li Y. Obstacles and coping strategies of CAR-T cell immunotherapy in solid tumors. Front Immunol (2021) 12:687822. doi: 10.3389/fimmu.2021.687822
67. Wilkie S, van Schalkwyk MCI, Hobbs S, Davies DM, van der Stegen SJC, Pereira ACP, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol (2012) 32:1059–70. doi: 10.1007/s10875-012-9689-9
68. Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell (2020) 38:473–88. doi: 10.1016/j.ccell.2020.07.005
69. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Trans Med (2021) 13:eabe7378. doi: 10.1126/scitranslmed.abe7378
70. Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med (2021) 13:eabd8836. doi: 10.1126/scitranslmed.abd8836
71. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-Based inhibitory chimeric antigen receptors (ICARs) divert off-target immunotherapy responses. Sci Transl Med (2013) 5:215ra172. doi: 10.1126/scitranslmed.3006597
72. Mayol K, Biajoux V, Marvel J, Balabanian K, Walzer T. Sequential desensitization of CXCR4 and S1P5 controls natural killer cell trafficking. Blood (2011) 118:4863–71. doi: 10.1182/blood-2011-06-362574
73. Wang AM, Ong XJ, D’Souza C, Neeson PJ, Zhu JJ. Combining chemotherapy with CAR-T cell therapy in treating solid tumors. Front Immunol (2023) 14:1140541. doi: 10.3389/fimmu.2023.1140541
74. Sasaya T, Kubo T, Murata K, Mizue Y, Sasaki K, Yanagawa J, et al. Cisplatin-induced HSF1-HSP90 axis enhances the expression of functional PD-L1 in oral squamous cell carcinoma. Cancer Med (2023) 12:4605–15. doi: 10.1002/cam4.5310
75. Guo S, Yao Y, Tang Y, Xin Z, Wu D, Ni C, et al. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Signal Transduct Target Ther (2023) 8:205. doi: 10.1038/s41392-023-01462-z
76. Nelson BE, Adashek JJ, Lin SH, Subbiah V. On target methods to induce abscopal phenomenon for off-target effects: from happenstance to happenings. Cancer Med (2023) 12:6451–65. doi: 10.1002/cam4.5454
77. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res (2018) 78:1031–43. doi: 10.1158/0008-5472.CAN-17-1788
78. Rassamegevanon T, Feindt L, Koi L, Müller J, Freudenberg R, Löck S, et al. Molecular response to combined molecular- and external radiotherapy in head and neck squamous cell carcinoma (HNSCC). Cancers (2021) 13:5595. doi: 10.3390/cancers13225595
79. Smith EL, Mailankody S, Staehr M, Wang X, Senechal B, Purdon TJ, et al. BCMA-targeted CAR T-cell therapy plus radiotherapy for the treatment of refractory myeloma reveals potential synergy. Cancer Immunol Res (2019) 7:1047–53. doi: 10.1158/2326-6066.CIR-18-0551
80. Yamazaki T, Sugita M, Martinet J, Boyer O, L. Galluzzi L, Guzman ML, et al. Boosting CAR T cell expansion and therapeutic activity with low-dose radiation therapy. Int J Radiat Oncol Biol Physics. (2020) 108:S158–9. doi: 10.1016/j.ijrobp.2020.07.920
81. Conry RM, LoBuglio AF, Wright M, Sumerel L, Pike MJ, Johanning F, et al. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res (1995) 55:1397–400.
82. Skipper J.C.A., Hendrickson R.C., Gulden P.H., Brichard V., Vanpel A., Chen Y., et al. An HLA-A2-restricted tyrosinase antigen on melanoma cells results from posttranslational modification and suggests a novel pathway for processing of membrane proteins. J Exp Med (1996) 183:527–34. doi: 10.1084/jem.183.2.527
83. Zarling AL, Ficarro SB, White FM, Shabanowitz J, Hunt DF, Engelhard VH. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J Exp Med (2000) 192:1755–62. doi: 10.1084/jem.192.12.1755
84. Reinhard K, Rengst B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science (2020) 367:446–53. doi: 10.1126/science.aay5967
85. Zhang C, Guo C, Li Y, Liu K, Zhao Q, Ouyang L. Identification of claudin-6 as a molecular biomarker in pan-cancer through multiple omics integrative analysis. Front Cell Dev Biol (2021) 9:726656. doi: 10.3389/fcell.2021.726656
86. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol (2009) 10:1185–92. doi: 10.1038/ni.1790
87. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313:1972–5. doi: 10.1126/science.1131078
88. Li K, Yuan Z, Lyu J, Ahn E, Davis SJ, Ahmed R, et al. PD-1 suppresses TCR-CD8 cooperativity during T-cell antigen recognition. Nat Commun (2021) 12:2746. doi: 10.1038/s41467-021-22965-9
89. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between programmed death-1 and programmed death ligand-1 promote tolerance by blocking the T cell receptor-induced stop signal. Nat Immunol (2009) 10:1185–92. doi: 10.1038/ni.1790
90. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126:3130–44. doi: 10.1172/JCI83092
91. Adusumilli PS, Zauderer MG, Rivière I, Solomon SB, Rusch VW, O’Cearbhaill RE, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti–PD-1 agent pembrolizumab. Cancer Discovery (2021) 11:2748–63. doi: 10.1158/2159-8290.CD-21-0407
92. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-Blocking ScFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol (2018) 36:847–56. doi: 10.1038/nbt.4195
93. Lee YH, Lee HJ, Kim HC, Lee Y, Nam SK, Hupperetz C, et al. PD-1 and TIGIT downregulation distinctly affect the effector and early memory phenotypes of CD19-targeting CAR T cells. Mol Ther (2022) 30:579–92. doi: 10.1016/j.ymthe.2021.10.004
94. Lin S, Cheng L, Ye W, Li S, Zheng D, Qin L, et al. Chimeric CTLA4-CD28-CD3z T cells potentiate antitumor activity against CD80/CD86-positive b cell malignancies. Front Immunol (2021) 12:642528. doi: 10.3389/fimmu.2021.642528
95. Zhou X, Cao H, Fang SY, Chow RD, Tang K, Majety M, et al. CTLA-4 tail fusion enhances CAR-T anti-tumour immunity. bioRxiv (2023). doi: 10.1101/2023.03.14.532655
96. Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 ligand-modified chimeric antigen receptor (CAR) T cells enhance antitumor function by eliciting an endogenous antitumor response. Cancer Cell (2019) 35:473–488.e6. doi: 10.1016/j.ccell.2019.02.006
97. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumour models. Cancer Discovery (2017) 7:1306–19. doi: 10.1158/2159-8290.CD-17-0263
98. Golubovskaya VM, Berahovich R, Xu Q, Zhou H, Xu S, Guan J, et al. GITR domain inside CAR co-stimulates activity of CAR-T cells against cancer. Front Biosci (Landmark Ed). (2018) 23:2245–54. doi: 10.2741/4703
99. Jin S, Wang Q, Wu H, Pang D, Xu S. Oncolytic viruses for triple negative breast cancer and beyond. biomark Res (2021) 9:71. doi: 10.1186/s40364-021-00318-4
100. Ma R, Li Z, Chiocca EA, Caligiuri MA, Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer. (2023) 9:122–39. doi: 10.1016/j.trecan.2022.10.003
101. Bell JC. Check and checkmate: battling cancer with multiplex immunotherapy. Mol Ther (2020) 28:1236–7. doi: 10.1016/j.ymthe.2020.04.013
102. Shaw RA, Porter CE, Watanabe N, Tanoue K, Sikora A, Gottschalk S, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther (2017) 25:2440–51. doi: 10.1016/j.ymthe.2017.09.010
103. Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Gooley T, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood (2019) 133:1876–87. doi: 10.1182/blood-2018-11-887067
104. Suryadevara CM, Desai R, Farber SH, Choi BD, Swartz AM, Shen SH, et al. Preventing lck activation in CAR T cells confers treg resistance but requires 4-1BB signaling for them to persist and treat solid tumors in nonlymphodepleted hosts. Clin Cancer Res (2019) 25:358–68. doi: 10.1158/1078-0432.CCR-18-1211
105. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol (2021) 18:2188–98. doi: 10.1038/s41423-021-00749-x
106. Beck JD, Reidenbach D, Salomon N, Sahin U, Türeci Ö., Vormehr M, et al. mRNA therapeutics in cancer immunotherapy. Mol Cancer. (2021) 20:69. doi: 10.1186/s12943-021-01348-0
107. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature (2016) 534:396–401. doi: 10.1038/nature18300
108. Wu X, Schmidt-Wolf IGH. An alternative source for allogeneic CAR T cells with a high safety profile. Front Immunol (2022) 13:913123. doi: 10.3389/fimmu.2022.913123
109. Guo X, Zheng H, Luo W, Zhang Q, Liu J, Yao K. 5T4-specific chimeric antigen receptor modification promotes the immune efficacy of cytokine-induced killer cells against nasopharyngeal carcinoma stem cell-like cells. Sci Rep (2017) 7:4859. doi: 10.1038/s41598-017-04756-9
110. Leuci V, Casucci M, Grignani G, Rotolo R, Rossotti U, Vigna E, et al. CD44v6 as innovative sarcoma target for CAR-redirected CIK cells. OncoImmunology (2018) 7:e1423167. doi: 10.1080/2162402X.2017.1423167
111. Merker M, Wagner J, Kreyenberg H, Heim C, Moser LM, Wels WS, et al. ERBB2-CAR-Engineered cytokine-induced killer cells exhibit both CAR-mediated and innate immunity against high-risk rhabdomyosarcoma. Front Immunol (2020) 11:581468. doi: 10.3389/fimmu.2020.581468
112. Cafri G, Gartner JJ, Zaks T, Hopson K, Levin N, Paria BC, et al. mRNA vaccine–induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest. (2020) 130:5976–88. doi: 10.1172/JCI134915
Keywords: CAR-T cell therapy, head and neck cancer, head and neck squamous cell carcinoma, immunotherapy, immune checkpoint inhibitors
Citation: Hu C, Liu M, Li Y, Zhao Y, Sharma A, Liu H and Schmidt-Wolf IGH (2023) Recent advances and future perspectives of CAR-T cell therapy in head and neck cancer. Front. Immunol. 14:1213716. doi: 10.3389/fimmu.2023.1213716
Received: 28 April 2023; Accepted: 12 June 2023;
Published: 29 June 2023.
Edited by:
Hamid Reza Mirzaei, Tehran University of Medical Sciences, IranReviewed by:
John Maher, King’s College London, United KingdomFatemeh Rahbarizadeh, Tarbiat Modares University, Iran
Copyright © 2023 Hu, Liu, Li, Zhao, Sharma, Liu and Schmidt-Wolf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haotian Liu, kevinliu712@wchscu.cn; Ingo G. H. Schmidt-Wolf, ingo.schmidt-wolf@ukbonn.de
†These authors have contributed equally to this work